![]()
ColabFold/AlphaFold2 Notebook#
ColabFold v1.5.3: AlphaFold2 using MMseqs2#
Easy to use protein structure and complex prediction using AlphaFold2 and Alphafold2-multimer. Sequence alignments/templates are generated through MMseqs2 and HHsearch. For more details, see bottom of the notebook, checkout the ColabFold GitHub and read our manuscript. Old versions: v1.4, v1.5.1, v1.5.2
News#
2023/07/31: The ColabFold MSA server is back to normal. It was using older DB (UniRef30 2202/PDB70 220313) from 27th ~8:30 AM CEST to 31st ~11:10 AM CEST.
2023/06/12: New databases! UniRef30 updated to 2023_02 and PDB to 230517. We now use PDB100 instead of PDB70 (see notes).
2023/06/12: We introduced a new default pairing strategy: Previously, for multimer predictions with more than 2 chains, we only pair if all sequences taxonomically match (“complete” pairing). The new default “greedy” strategy pairs any taxonomically matching subsets.
# @title Input protein sequence(s), then hit `Runtime` -> `Run all` { display-mode: "form" }
from google.colab import files
import os
import re
import hashlib
import random
from sys import version_info
python_version = f"{version_info.major}.{version_info.minor}"
def add_hash(x,y):
return x+"_"+hashlib.sha1(y.encode()).hexdigest()[:5]
query_sequence = 'PIAQIHILEGRSDEQKETLIREVSEAISRSLDAPLTSVRVIITEMAKGHFGIGGELASK' #@param {type:"string"}
#@markdown - Use `:` to specify inter-protein chainbreaks for **modeling complexes** (supports homo- and hetro-oligomers). For example **PI...SK:PI...SK** for a homodimer
jobname = 'test' #@param {type:"string"}
# number of models to use
num_relax = 0 #@param [0, 1, 5] {type:"raw"}
#@markdown - specify how many of the top ranked structures to relax using amber
template_mode = "none" #@param ["none", "pdb100","custom"]
#@markdown - `none` = no template information is used. `pdb100` = detect templates in pdb100 (see [notes](#pdb100)). `custom` - upload and search own templates (PDB or mmCIF format, see [notes](#custom_templates))
use_amber = num_relax > 0
# remove whitespaces
query_sequence = "".join(query_sequence.split())
basejobname = "".join(jobname.split())
basejobname = re.sub(r'\W+', '', basejobname)
jobname = add_hash(basejobname, query_sequence)
# check if directory with jobname exists
def check(folder):
if os.path.exists(folder):
return False
else:
return True
if not check(jobname):
n = 0
while not check(f"{jobname}_{n}"): n += 1
jobname = f"{jobname}_{n}"
# make directory to save results
os.makedirs(jobname, exist_ok=True)
# save queries
queries_path = os.path.join(jobname, f"{jobname}.csv")
with open(queries_path, "w") as text_file:
text_file.write(f"id,sequence\n{jobname},{query_sequence}")
if template_mode == "pdb100":
use_templates = True
custom_template_path = None
elif template_mode == "custom":
custom_template_path = os.path.join(jobname,f"template")
os.makedirs(custom_template_path, exist_ok=True)
uploaded = files.upload()
use_templates = True
for fn in uploaded.keys():
os.rename(fn,os.path.join(custom_template_path,fn))
else:
custom_template_path = None
use_templates = False
print("jobname",jobname)
print("sequence",query_sequence)
print("length",len(query_sequence.replace(":","")))
jobname test_a5e17
sequence PIAQIHILEGRSDEQKETLIREVSEAISRSLDAPLTSVRVIITEMAKGHFGIGGELASK
length 59
# @title Install dependencies { display-mode: "form" }
%%time
import os
USE_AMBER = use_amber
USE_TEMPLATES = use_templates
PYTHON_VERSION = python_version
if not os.path.isfile("COLABFOLD_READY"):
print("installing colabfold...")
os.system("pip install -q --no-warn-conflicts 'colabfold[alphafold-minus-jax] @ git+https://github.com/sokrypton/ColabFold'")
os.system("pip install --upgrade dm-haiku")
os.system("ln -s /usr/local/lib/python3.*/dist-packages/colabfold colabfold")
os.system("ln -s /usr/local/lib/python3.*/dist-packages/alphafold alphafold")
# patch for jax > 0.3.25
os.system("sed -i 's/weights = jax.nn.softmax(logits)/logits=jnp.clip(logits,-1e8,1e8);weights=jax.nn.softmax(logits)/g' alphafold/model/modules.py")
os.system("touch COLABFOLD_READY")
if USE_AMBER or USE_TEMPLATES:
if not os.path.isfile("CONDA_READY"):
print("installing conda...")
os.system("wget -qnc https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh")
os.system("bash Mambaforge-Linux-x86_64.sh -bfp /usr/local")
os.system("mamba config --set auto_update_conda false")
os.system("touch CONDA_READY")
if USE_TEMPLATES and not os.path.isfile("HH_READY") and USE_AMBER and not os.path.isfile("AMBER_READY"):
print("installing hhsuite and amber...")
os.system(f"mamba install -y -c conda-forge -c bioconda kalign2=2.04 hhsuite=3.3.0 openmm=7.7.0 python='{PYTHON_VERSION}' pdbfixer")
os.system("touch HH_READY")
os.system("touch AMBER_READY")
else:
if USE_TEMPLATES and not os.path.isfile("HH_READY"):
print("installing hhsuite...")
os.system(f"mamba install -y -c conda-forge -c bioconda kalign2=2.04 hhsuite=3.3.0 python='{PYTHON_VERSION}'")
os.system("touch HH_READY")
if USE_AMBER and not os.path.isfile("AMBER_READY"):
print("installing amber...")
os.system(f"mamba install -y -c conda-forge openmm=7.7.0 python='{PYTHON_VERSION}' pdbfixer")
os.system("touch AMBER_READY")
installing colabfold...
CPU times: user 114 ms, sys: 27.2 ms, total: 142 ms
Wall time: 42.9 s
#@markdown ### MSA options (custom MSA upload, single sequence, pairing mode)
msa_mode = "mmseqs2_uniref_env" #@param ["mmseqs2_uniref_env", "mmseqs2_uniref","single_sequence","custom"]
pair_mode = "unpaired_paired" #@param ["unpaired_paired","paired","unpaired"] {type:"string"}
#@markdown - "unpaired_paired" = pair sequences from same species + unpaired MSA, "unpaired" = seperate MSA for each chain, "paired" - only use paired sequences.
# decide which a3m to use
if "mmseqs2" in msa_mode:
a3m_file = os.path.join(jobname,f"{jobname}.a3m")
elif msa_mode == "custom":
a3m_file = os.path.join(jobname,f"{jobname}.custom.a3m")
if not os.path.isfile(a3m_file):
custom_msa_dict = files.upload()
custom_msa = list(custom_msa_dict.keys())[0]
header = 0
import fileinput
for line in fileinput.FileInput(custom_msa,inplace=1):
if line.startswith(">"):
header = header + 1
if not line.rstrip():
continue
if line.startswith(">") == False and header == 1:
query_sequence = line.rstrip()
print(line, end='')
os.rename(custom_msa, a3m_file)
queries_path=a3m_file
print(f"moving {custom_msa} to {a3m_file}")
else:
a3m_file = os.path.join(jobname,f"{jobname}.single_sequence.a3m")
with open(a3m_file, "w") as text_file:
text_file.write(">1\n%s" % query_sequence)
# @title { display-mode: "form" }
#@markdown ### Advanced settings
model_type = "auto" #@param ["auto", "alphafold2_ptm", "alphafold2_multimer_v1", "alphafold2_multimer_v2", "alphafold2_multimer_v3"]
#@markdown - if `auto` selected, will use `alphafold2_ptm` for monomer prediction and `alphafold2_multimer_v3` for complex prediction.
#@markdown Any of the mode_types can be used (regardless if input is monomer or complex).
num_recycles = "3" #@param ["auto", "0", "1", "3", "6", "12", "24", "48"]
#@markdown - if `auto` selected, will use `num_recycles=20` if `model_type=alphafold2_multimer_v3`, else `num_recycles=3` .
recycle_early_stop_tolerance = "auto" #@param ["auto", "0.0", "0.5", "1.0"]
#@markdown - if `auto` selected, will use `tol=0.5` if `model_type=alphafold2_multimer_v3` else `tol=0.0`.
relax_max_iterations = 200 #@param [0, 200, 2000] {type:"raw"}
#@markdown - max amber relax iterations, `0` = unlimited (AlphaFold2 default, can take very long)
pairing_strategy = "greedy" #@param ["greedy", "complete"] {type:"string"}
#@markdown - `greedy` = pair any taxonomically matching subsets, `complete` = all sequences have to match in one line.
#@markdown #### Sample settings
#@markdown - enable dropouts and increase number of seeds to sample predictions from uncertainty of the model.
#@markdown - decrease `max_msa` to increase uncertainity
max_msa = "auto" #@param ["auto", "512:1024", "256:512", "64:128", "32:64", "16:32"]
num_seeds = 1 #@param [1,2,4,8,16] {type:"raw"}
use_dropout = False #@param {type:"boolean"}
num_recycles = None if num_recycles == "auto" else int(num_recycles)
recycle_early_stop_tolerance = None if recycle_early_stop_tolerance == "auto" else float(recycle_early_stop_tolerance)
if max_msa == "auto": max_msa = None
#@markdown #### Save settings
save_all = False #@param {type:"boolean"}
save_recycles = False #@param {type:"boolean"}
save_to_google_drive = False #@param {type:"boolean"}
#@markdown - if the save_to_google_drive option was selected, the result zip will be uploaded to your Google Drive
dpi = 200 #@param {type:"integer"}
#@markdown - set dpi for image resolution
if save_to_google_drive:
from pydrive.drive import GoogleDrive
from pydrive.auth import GoogleAuth
from google.colab import auth
from oauth2client.client import GoogleCredentials
auth.authenticate_user()
gauth = GoogleAuth()
gauth.credentials = GoogleCredentials.get_application_default()
drive = GoogleDrive(gauth)
print("You are logged into Google Drive and are good to go!")
#@markdown Don't forget to hit `Runtime` -> `Run all` after updating the form.
#@title Run Prediction
display_images = True #@param {type:"boolean"}
import sys
import warnings
warnings.simplefilter(action='ignore', category=FutureWarning)
from Bio import BiopythonDeprecationWarning
warnings.simplefilter(action='ignore', category=BiopythonDeprecationWarning)
from pathlib import Path
from colabfold.download import download_alphafold_params, default_data_dir
from colabfold.utils import setup_logging
from colabfold.batch import get_queries, run, set_model_type
from colabfold.plot import plot_msa_v2
import os
import numpy as np
try:
K80_chk = os.popen('nvidia-smi | grep "Tesla K80" | wc -l').read()
except:
K80_chk = "0"
pass
if "1" in K80_chk:
print("WARNING: found GPU Tesla K80: limited to total length < 1000")
if "TF_FORCE_UNIFIED_MEMORY" in os.environ:
del os.environ["TF_FORCE_UNIFIED_MEMORY"]
if "XLA_PYTHON_CLIENT_MEM_FRACTION" in os.environ:
del os.environ["XLA_PYTHON_CLIENT_MEM_FRACTION"]
from colabfold.colabfold import plot_protein
from pathlib import Path
import matplotlib.pyplot as plt
# For some reason we need that to get pdbfixer to import
if use_amber and f"/usr/local/lib/python{python_version}/site-packages/" not in sys.path:
sys.path.insert(0, f"/usr/local/lib/python{python_version}/site-packages/")
def input_features_callback(input_features):
if display_images:
plot_msa_v2(input_features)
plt.show()
plt.close()
def prediction_callback(protein_obj, length,
prediction_result, input_features, mode):
model_name, relaxed = mode
if not relaxed:
if display_images:
fig = plot_protein(protein_obj, Ls=length, dpi=150)
plt.show()
plt.close()
result_dir = jobname
log_filename = os.path.join(jobname,"log.txt")
setup_logging(Path(log_filename))
queries, is_complex = get_queries(queries_path)
model_type = set_model_type(is_complex, model_type)
if "multimer" in model_type and max_msa is not None:
use_cluster_profile = False
else:
use_cluster_profile = True
download_alphafold_params(model_type, Path("."))
results = run(
queries=queries,
result_dir=result_dir,
use_templates=use_templates,
custom_template_path=custom_template_path,
num_relax=num_relax,
msa_mode=msa_mode,
model_type=model_type,
num_models=5,
num_recycles=num_recycles,
relax_max_iterations=relax_max_iterations,
recycle_early_stop_tolerance=recycle_early_stop_tolerance,
num_seeds=num_seeds,
use_dropout=use_dropout,
model_order=[1,2,3,4,5],
is_complex=is_complex,
data_dir=Path("."),
keep_existing_results=False,
rank_by="auto",
pair_mode=pair_mode,
pairing_strategy=pairing_strategy,
stop_at_score=float(100),
prediction_callback=prediction_callback,
dpi=dpi,
zip_results=False,
save_all=save_all,
max_msa=max_msa,
use_cluster_profile=use_cluster_profile,
input_features_callback=input_features_callback,
save_recycles=save_recycles,
user_agent="colabfold/google-colab-main",
)
results_zip = f"{jobname}.result.zip"
os.system(f"zip -r {results_zip} {jobname}")
Downloading alphafold2 weights to .: 100%|██████████| 3.47G/3.47G [02:40<00:00, 23.2MB/s]
2023-12-07 22:02:42,821 Unable to initialize backend 'rocm': NOT_FOUND: Could not find registered platform with name: "rocm". Available platform names are: CUDA
2023-12-07 22:02:42,823 Unable to initialize backend 'tpu': INTERNAL: Failed to open libtpu.so: libtpu.so: cannot open shared object file: No such file or directory
2023-12-07 22:02:44,726 Running on GPU
2023-12-07 22:02:44,904 Found 4 citations for tools or databases
2023-12-07 22:02:44,904 Query 1/1: test_a5e17 (length 59)
COMPLETE: 100%|██████████| 150/150 [elapsed: 00:01 remaining: 00:00]
2023-12-07 22:02:47,182 Setting max_seq=512, max_extra_seq=5120
---------------------------------------------------------------------------
KeyboardInterrupt Traceback (most recent call last)
<ipython-input-5-06098a2c69e9> in <cell line: 65>()
63
64 download_alphafold_params(model_type, Path("."))
---> 65 results = run(
66 queries=queries,
67 result_dir=result_dir,
/content/colabfold/batch.py in run(queries, result_dir, num_models, is_complex, num_recycles, recycle_early_stop_tolerance, model_order, num_ensemble, model_type, msa_mode, use_templates, custom_template_path, num_relax, relax_max_iterations, relax_tolerance, relax_stiffness, relax_max_outer_iterations, keep_existing_results, rank_by, pair_mode, pairing_strategy, data_dir, host_url, user_agent, random_seed, num_seeds, recompile_padding, zip_results, prediction_callback, save_single_representations, save_pair_representations, save_all, save_recycles, use_dropout, use_gpu_relax, stop_at_score, dpi, max_seq, max_extra_seq, pdb_hit_file, local_pdb_path, use_cluster_profile, feature_dict_callback, **kwargs)
1568 first_job = False
1569
-> 1570 results = predict_structure(
1571 prefix=jobname,
1572 result_dir=result_dir,
/content/colabfold/batch.py in predict_structure(prefix, result_dir, feature_dict, is_complex, use_templates, sequences_lengths, pad_len, model_type, model_runner_and_params, num_relax, relax_max_iterations, relax_tolerance, relax_stiffness, relax_max_outer_iterations, rank_by, random_seed, num_seeds, stop_at_score, prediction_callback, use_gpu_relax, save_all, save_single_representations, save_pair_representations, save_recycles)
419 # predict
420 result, recycles = \
--> 421 model_runner.predict(input_features,
422 random_seed=seed,
423 return_representations=return_representations,
/content/alphafold/model/model.py in predict(self, feat, random_seed, return_representations, callback)
183 # run
184 key, sub_key = jax.random.split(key)
--> 185 result, prev = run(sub_key, sub_feat, prev)
186
187 if return_representations:
/content/alphafold/model/model.py in run(key, feat, prev)
163 x[k] = np.asarray(v,np.float16)
164 return x
--> 165 result = _jnp_to_np(self.apply(self.params, key, {**feat, "prev":prev}))
166 prev = result.pop("prev")
167 return result, prev
[... skipping hidden 1 frame]
/usr/local/lib/python3.10/dist-packages/jax/_src/pjit.py in cache_miss(*args, **kwargs)
254 @api_boundary
255 def cache_miss(*args, **kwargs):
--> 256 outs, out_flat, out_tree, args_flat, jaxpr = _python_pjit_helper(
257 fun, infer_params_fn, *args, **kwargs)
258 executable = _read_most_recent_pjit_call_executable(jaxpr)
/usr/local/lib/python3.10/dist-packages/jax/_src/pjit.py in _python_pjit_helper(fun, infer_params_fn, *args, **kwargs)
165 dispatch.check_arg(arg)
166 try:
--> 167 out_flat = pjit_p.bind(*args_flat, **params)
168 except pxla.DeviceAssignmentMismatchError as e:
169 fails, = e.args
/usr/local/lib/python3.10/dist-packages/jax/_src/core.py in bind(self, *args, **params)
2654 top_trace = (top_trace if not axis_main or axis_main.level < top_trace.level
2655 else axis_main.with_cur_sublevel())
-> 2656 return self.bind_with_trace(top_trace, args, params)
2657
2658
/usr/local/lib/python3.10/dist-packages/jax/_src/core.py in bind_with_trace(self, trace, args, params)
386
387 def bind_with_trace(self, trace, args, params):
--> 388 out = trace.process_primitive(self, map(trace.full_raise, args), params)
389 return map(full_lower, out) if self.multiple_results else full_lower(out)
390
/usr/local/lib/python3.10/dist-packages/jax/_src/core.py in process_primitive(self, primitive, tracers, params)
866
867 def process_primitive(self, primitive, tracers, params):
--> 868 return primitive.impl(*tracers, **params)
869
870 def process_call(self, primitive, f, tracers, params):
/usr/local/lib/python3.10/dist-packages/jax/_src/pjit.py in _pjit_call_impl(jaxpr, in_shardings, out_shardings, resource_env, donated_invars, name, keep_unused, inline, *args)
1210 has_explicit_sharding = _pjit_explicit_sharding(
1211 in_shardings, out_shardings, None, None)
-> 1212 return xc._xla.pjit(name, f, call_impl_cache_miss, [], [], donated_argnums,
1213 tree_util.dispatch_registry,
1214 _get_cpp_global_cache(has_explicit_sharding))(*args)
/usr/local/lib/python3.10/dist-packages/jax/_src/pjit.py in call_impl_cache_miss(*args_, **kwargs_)
1194 donated_invars, name, keep_unused, inline):
1195 def call_impl_cache_miss(*args_, **kwargs_):
-> 1196 out_flat, compiled = _pjit_call_impl_python(
1197 *args, jaxpr=jaxpr, in_shardings=in_shardings,
1198 out_shardings=out_shardings, resource_env=resource_env,
/usr/local/lib/python3.10/dist-packages/jax/_src/pjit.py in _pjit_call_impl_python(jaxpr, in_shardings, out_shardings, resource_env, donated_invars, name, keep_unused, inline, *args)
1127 resource_env.physical_mesh if resource_env is not None else None)
1128
-> 1129 compiled = _pjit_lower(
1130 jaxpr, in_shardings, out_shardings, resource_env,
1131 donated_invars, name, keep_unused, inline,
/usr/local/lib/python3.10/dist-packages/jax/_src/pjit.py in _pjit_lower(jaxpr, in_shardings, out_shardings, *args, **kwargs)
1258 in_shardings = SameDeviceAssignmentTuple(tuple(in_shardings), da)
1259 out_shardings = SameDeviceAssignmentTuple(tuple(out_shardings), da)
-> 1260 return _pjit_lower_cached(jaxpr, in_shardings, out_shardings, *args, **kwargs)
1261
1262
/usr/local/lib/python3.10/dist-packages/jax/_src/pjit.py in _pjit_lower_cached(jaxpr, sdat_in_shardings, sdat_out_shardings, resource_env, donated_invars, name, keep_unused, inline, lowering_parameters)
1297 lowering_parameters=lowering_parameters)
1298 else:
-> 1299 return pxla.lower_sharding_computation(
1300 jaxpr, api_name, name, in_shardings, out_shardings,
1301 tuple(donated_invars), tuple(jaxpr.in_avals),
/usr/local/lib/python3.10/dist-packages/jax/_src/profiler.py in wrapper(*args, **kwargs)
338 def wrapper(*args, **kwargs):
339 with TraceAnnotation(name, **decorator_kwargs):
--> 340 return func(*args, **kwargs)
341 return wrapper
342 return wrapper
/usr/local/lib/python3.10/dist-packages/jax/_src/interpreters/pxla.py in lower_sharding_computation(fun_or_jaxpr, api_name, fun_name, in_shardings, out_shardings, donated_invars, global_in_avals, keep_unused, inline, devices_from_context, lowering_parameters)
2029 semantic_out_shardings = SemanticallyEqualShardings(out_shardings)
2030 (module, keepalive, host_callbacks, unordered_effects, ordered_effects,
-> 2031 nreps, tuple_args, shape_poly_state) = _cached_lowering_to_hlo(
2032 closed_jaxpr, api_name, fun_name, backend, semantic_in_shardings,
2033 semantic_out_shardings, da_object,
/usr/local/lib/python3.10/dist-packages/jax/_src/interpreters/pxla.py in _cached_lowering_to_hlo(closed_jaxpr, api_name, fun_name, backend, semantic_in_shardings, semantic_out_shardings, da_object, donated_invars, name_stack, all_default_mem_kind, lowering_parameters)
1830 "Finished jaxpr to MLIR module conversion {fun_name} in {elapsed_time} sec",
1831 fun_name=str(name_stack), event=dispatch.JAXPR_TO_MLIR_MODULE_EVENT):
-> 1832 lowering_result = mlir.lower_jaxpr_to_module(
1833 module_name,
1834 closed_jaxpr,
/usr/local/lib/python3.10/dist-packages/jax/_src/interpreters/mlir.py in lower_jaxpr_to_module(module_name, jaxpr, ordered_effects, backend_or_name, platforms, axis_context, name_stack, donated_args, replicated_args, arg_shardings, result_shardings, arg_names, result_names, num_replicas, num_partitions, all_default_mem_kind, lowering_parameters)
804 attrs["mhlo.num_partitions"] = i32_attr(num_partitions)
805 replace_tokens_with_dummy = lowering_parameters.replace_tokens_with_dummy
--> 806 lower_jaxpr_to_fun(
807 ctx, "main", jaxpr, ordered_effects, public=True,
808 create_tokens=replace_tokens_with_dummy,
/usr/local/lib/python3.10/dist-packages/jax/_src/interpreters/mlir.py in lower_jaxpr_to_fun(ctx, name, jaxpr, effects, create_tokens, public, replace_tokens_with_dummy, replicated_args, arg_shardings, result_shardings, use_sharding_annotations, input_output_aliases, num_output_tokens, api_name, arg_names, result_names, arg_memory_kinds, result_memory_kinds)
1211 callee_name_stack = ctx.name_stack.extend(util.wrap_name(name, api_name))
1212 consts = [ir_constants(xla.canonicalize_dtype(x)) for x in jaxpr.consts]
-> 1213 out_vals, tokens_out = jaxpr_subcomp(
1214 ctx.replace(name_stack=callee_name_stack), jaxpr.jaxpr, tokens_in,
1215 consts, *args, dim_var_values=dim_var_values)
/usr/local/lib/python3.10/dist-packages/jax/_src/interpreters/mlir.py in jaxpr_subcomp(ctx, jaxpr, tokens, consts, dim_var_values, *args)
1429 if len(ctx.platforms) == 1:
1430 # Classic, single-platform lowering
-> 1431 ans = rule(rule_ctx, *rule_inputs, **eqn.params)
1432 else:
1433 ans = lower_multi_platform(rule_ctx, str(eqn), rules,
/usr/local/lib/python3.10/dist-packages/jax/_src/interpreters/mlir.py in f_lowered(ctx, *args, **params)
1626 # TODO(frostig,mattjj): check ctx.avals_out against jaxpr avals out?
1627
-> 1628 out, tokens = jaxpr_subcomp(
1629 ctx.module_context, jaxpr, ctx.tokens_in, _ir_consts(consts),
1630 *map(wrap_singleton_ir_values, args), dim_var_values=ctx.dim_var_values)
/usr/local/lib/python3.10/dist-packages/jax/_src/interpreters/mlir.py in jaxpr_subcomp(ctx, jaxpr, tokens, consts, dim_var_values, *args)
1429 if len(ctx.platforms) == 1:
1430 # Classic, single-platform lowering
-> 1431 ans = rule(rule_ctx, *rule_inputs, **eqn.params)
1432 else:
1433 ans = lower_multi_platform(rule_ctx, str(eqn), rules,
/usr/local/lib/python3.10/dist-packages/jax/_src/lax/control_flow/loops.py in _while_lowering(ctx, cond_jaxpr, body_jaxpr, cond_nconsts, body_nconsts, *args)
1670 body_consts = [mlir.ir_constants(xla.canonicalize_dtype(x))
1671 for x in body_jaxpr.consts]
-> 1672 new_z, tokens_out = mlir.jaxpr_subcomp(body_ctx, body_jaxpr.jaxpr,
1673 tokens_in, body_consts, *(y + z), dim_var_values=ctx.dim_var_values)
1674 out_tokens = [tokens_out.get(eff) for eff in body_effects]
/usr/local/lib/python3.10/dist-packages/jax/_src/interpreters/mlir.py in jaxpr_subcomp(ctx, jaxpr, tokens, consts, dim_var_values, *args)
1429 if len(ctx.platforms) == 1:
1430 # Classic, single-platform lowering
-> 1431 ans = rule(rule_ctx, *rule_inputs, **eqn.params)
1432 else:
1433 ans = lower_multi_platform(rule_ctx, str(eqn), rules,
/usr/local/lib/python3.10/dist-packages/jax/_src/interpreters/mlir.py in f_lowered(ctx, *args, **params)
1626 # TODO(frostig,mattjj): check ctx.avals_out against jaxpr avals out?
1627
-> 1628 out, tokens = jaxpr_subcomp(
1629 ctx.module_context, jaxpr, ctx.tokens_in, _ir_consts(consts),
1630 *map(wrap_singleton_ir_values, args), dim_var_values=ctx.dim_var_values)
/usr/local/lib/python3.10/dist-packages/jax/_src/interpreters/mlir.py in jaxpr_subcomp(ctx, jaxpr, tokens, consts, dim_var_values, *args)
1429 if len(ctx.platforms) == 1:
1430 # Classic, single-platform lowering
-> 1431 ans = rule(rule_ctx, *rule_inputs, **eqn.params)
1432 else:
1433 ans = lower_multi_platform(rule_ctx, str(eqn), rules,
/usr/local/lib/python3.10/dist-packages/jax/_src/lax/control_flow/loops.py in _while_lowering(ctx, cond_jaxpr, body_jaxpr, cond_nconsts, body_nconsts, *args)
1632 mlir.ir_constants(xla.canonicalize_dtype(x)) for x in cond_jaxpr.consts
1633 ]
-> 1634 ((pred,),), _ = mlir.jaxpr_subcomp(
1635 cond_ctx,
1636 cond_jaxpr.jaxpr,
/usr/local/lib/python3.10/dist-packages/jax/_src/interpreters/mlir.py in jaxpr_subcomp(ctx, jaxpr, tokens, consts, dim_var_values, *args)
1313 return func_op
1314
-> 1315 def jaxpr_subcomp(ctx: ModuleContext, jaxpr: core.Jaxpr,
1316 tokens: TokenSet,
1317 consts: Sequence[Sequence[ir.Value]],
KeyboardInterrupt:
#@title Display 3D structure {run: "auto"}
import py3Dmol
import glob
import matplotlib.pyplot as plt
from colabfold.colabfold import plot_plddt_legend
from colabfold.colabfold import pymol_color_list, alphabet_list
rank_num = 1 #@param ["1", "2", "3", "4", "5"] {type:"raw"}
color = "lDDT" #@param ["chain", "lDDT", "rainbow"]
show_sidechains = False #@param {type:"boolean"}
show_mainchains = False #@param {type:"boolean"}
tag = results["rank"][0][rank_num - 1]
jobname_prefix = ".custom" if msa_mode == "custom" else ""
pdb_filename = f"{jobname}/{jobname}{jobname_prefix}_unrelaxed_{tag}.pdb"
pdb_file = glob.glob(pdb_filename)
def show_pdb(rank_num=1, show_sidechains=False, show_mainchains=False, color="lDDT"):
model_name = f"rank_{rank_num}"
view = py3Dmol.view(js='https://3dmol.org/build/3Dmol.js',)
view.addModel(open(pdb_file[0],'r').read(),'pdb')
if color == "lDDT":
view.setStyle({'cartoon': {'colorscheme': {'prop':'b','gradient': 'roygb','min':50,'max':90}}})
elif color == "rainbow":
view.setStyle({'cartoon': {'color':'spectrum'}})
elif color == "chain":
chains = len(queries[0][1]) + 1 if is_complex else 1
for n,chain,color in zip(range(chains),alphabet_list,pymol_color_list):
view.setStyle({'chain':chain},{'cartoon': {'color':color}})
if show_sidechains:
BB = ['C','O','N']
view.addStyle({'and':[{'resn':["GLY","PRO"],'invert':True},{'atom':BB,'invert':True}]},
{'stick':{'colorscheme':f"WhiteCarbon",'radius':0.3}})
view.addStyle({'and':[{'resn':"GLY"},{'atom':'CA'}]},
{'sphere':{'colorscheme':f"WhiteCarbon",'radius':0.3}})
view.addStyle({'and':[{'resn':"PRO"},{'atom':['C','O'],'invert':True}]},
{'stick':{'colorscheme':f"WhiteCarbon",'radius':0.3}})
if show_mainchains:
BB = ['C','O','N','CA']
view.addStyle({'atom':BB},{'stick':{'colorscheme':f"WhiteCarbon",'radius':0.3}})
view.zoomTo()
return view
show_pdb(rank_num, show_sidechains, show_mainchains, color).show()
if color == "lDDT":
plot_plddt_legend().show()
#@title Plots {run: "auto"}
from IPython.display import display, HTML
import base64
from html import escape
# see: https://stackoverflow.com/a/53688522
def image_to_data_url(filename):
ext = filename.split('.')[-1]
prefix = f'data:image/{ext};base64,'
with open(filename, 'rb') as f:
img = f.read()
return prefix + base64.b64encode(img).decode('utf-8')
pae = image_to_data_url(os.path.join(jobname,f"{jobname}{jobname_prefix}_pae.png"))
cov = image_to_data_url(os.path.join(jobname,f"{jobname}{jobname_prefix}_coverage.png"))
plddt = image_to_data_url(os.path.join(jobname,f"{jobname}{jobname_prefix}_plddt.png"))
display(HTML(f"""
<style>
img {{
float:left;
}}
.full {{
max-width:100%;
}}
.half {{
max-width:50%;
}}
@media (max-width:640px) {{
.half {{
max-width:100%;
}}
}}
</style>
<div style="max-width:90%; padding:2em;">
<h1>Plots for {escape(jobname)}</h1>
<img src="{pae}" class="full" />
<img src="{cov}" class="half" />
<img src="{plddt}" class="half" />
</div>
"""))
#@title Package and download results
#@markdown If you are having issues downloading the result archive, try disabling your adblocker and run this cell again. If that fails click on the little folder icon to the left, navigate to file: `jobname.result.zip`, right-click and select \"Download\" (see [screenshot](https://pbs.twimg.com/media/E6wRW2lWUAEOuoe?format=jpg&name=small)).
if msa_mode == "custom":
print("Don't forget to cite your custom MSA generation method.")
files.download(f"{jobname}.result.zip")
if save_to_google_drive == True and drive:
uploaded = drive.CreateFile({'title': f"{jobname}.result.zip"})
uploaded.SetContentFile(f"{jobname}.result.zip")
uploaded.Upload()
print(f"Uploaded {jobname}.result.zip to Google Drive with ID {uploaded.get('id')}")
Instructions #
Quick start
Paste your protein sequence(s) in the input field.
Press “Runtime” -> “Run all”.
The pipeline consists of 5 steps. The currently running step is indicated by a circle with a stop sign next to it.
Result zip file contents
PDB formatted structures sorted by avg. pLDDT and complexes are sorted by pTMscore. (unrelaxed and relaxed if
use_amberis enabled).Plots of the model quality.
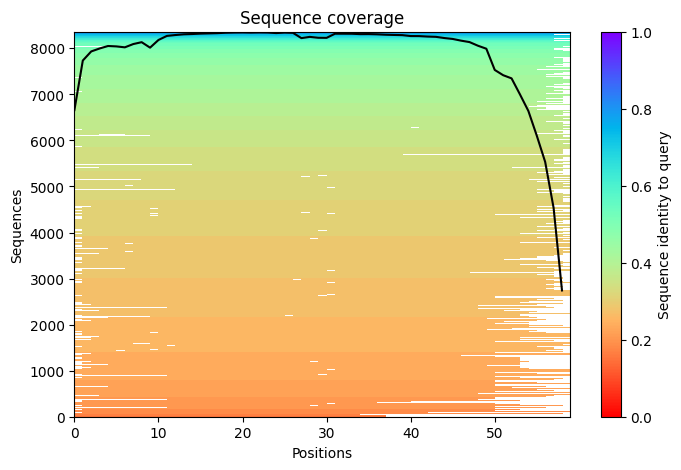
Plots of the MSA coverage.
Parameter log file.
A3M formatted input MSA.
A
predicted_aligned_error_v1.jsonusing AlphaFold-DB’s format and ascores.jsonfor each model which contains an array (list of lists) for PAE, a list with the average pLDDT and the pTMscore.BibTeX file with citations for all used tools and databases.
At the end of the job a download modal box will pop up with a jobname.result.zip file. Additionally, if the save_to_google_drive option was selected, the jobname.result.zip will be uploaded to your Google Drive.
MSA generation for complexes
For the complex prediction we use unpaired and paired MSAs. Unpaired MSA is generated the same way as for the protein structures prediction by searching the UniRef100 and environmental sequences three iterations each.
The paired MSA is generated by searching the UniRef100 database and pairing the best hits sharing the same NCBI taxonomic identifier (=species or sub-species). We only pair sequences if all of the query sequences are present for the respective taxonomic identifier.
Using a custom MSA as input
To predict the structure with a custom MSA (A3M formatted): (1) Change the msa_mode: to “custom”, (2) Wait for an upload box to appear at the end of the “MSA options …” box. Upload your A3M. The first fasta entry of the A3M must be the query sequence without gaps.
It is also possilbe to proide custom MSAs for complex predictions. Read more about the format here.
As an alternative for MSA generation the HHblits Toolkit server can be used. After submitting your query, click “Query Template MSA” -> “Download Full A3M”. Download the A3M file and upload it in this notebook.
As of 23/06/08, we have transitioned from using the PDB70 to a 100% clustered PDB, the PDB100. The construction methodology of PDB100 differs from that of PDB70.
The PDB70 was constructed by running each PDB70 representative sequence through HHblits against the Uniclust30. On the other hand, the PDB100 is built by searching each PDB100 representative structure with Foldseek against the AlphaFold Database.
To maintain compatibility with older Notebook versions and local installations, the generated files and API responses will continue to be named “PDB70”, even though we’re now using the PDB100.
To predict the structure with a custom template (PDB or mmCIF formatted): (1) change the template_mode to “custom” in the execute cell and (2) wait for an upload box to appear at the end of the “Input Protein” box. Select and upload your templates (multiple choices are possible).
Templates must follow the four letter PDB naming with lower case letters.
Templates in mmCIF format must contain
_entity_poly_seq. An error is thrown if this field is not present. The field_pdbx_audit_revision_history.revision_dateis automatically generated if it is not present.Templates in PDB format are automatically converted to the mmCIF format.
_entity_poly_seqand_pdbx_audit_revision_history.revision_dateare automatically generated.
If you encounter problems, please report them to this issue.
Comparison to the full AlphaFold2 and AlphaFold2 Colab
This notebook replaces the homology detection and MSA pairing of AlphaFold2 with MMseqs2. For a comparison against the AlphaFold2 Colab and the full AlphaFold2 system read our paper.
Troubleshooting
Check that the runtime type is set to GPU at “Runtime” -> “Change runtime type”.
Try to restart the session “Runtime” -> “Factory reset runtime”.
Check your input sequence.
Known issues
Google Colab assigns different types of GPUs with varying amount of memory. Some might not have enough memory to predict the structure for a long sequence.
Your browser can block the pop-up for downloading the result file. You can choose the
save_to_google_driveoption to upload to Google Drive instead or manually download the result file: Click on the little folder icon to the left, navigate to file:jobname.result.zip, right-click and select “Download” (see screenshot).
Limitations
Computing resources: Our MMseqs2 API can handle ~20-50k requests per day.
MSAs: MMseqs2 is very precise and sensitive but might find less hits compared to HHblits/HMMer searched against BFD or MGnify.
We recommend to additionally use the full AlphaFold2 pipeline.
Description of the plots
Number of sequences per position - We want to see at least 30 sequences per position, for best performance, ideally 100 sequences.
Predicted lDDT per position - model confidence (out of 100) at each position. The higher the better.
Predicted Alignment Error - For homooligomers, this could be a useful metric to assess how confident the model is about the interface. The lower the better.
Bugs
If you encounter any bugs, please report the issue to sokrypton/ColabFold#issues
License
The source code of ColabFold is licensed under MIT. Additionally, this notebook uses the AlphaFold2 source code and its parameters licensed under Apache 2.0 and CC BY 4.0 respectively. Read more about the AlphaFold license here.
Acknowledgments
We thank the AlphaFold team for developing an excellent model and open sourcing the software.
KOBIC and Söding Lab for providing the computational resources for the MMseqs2 MSA server.
Richard Evans for helping to benchmark the ColabFold’s Alphafold-multimer support.
David Koes for his awesome py3Dmol plugin, without whom these notebooks would be quite boring!
Do-Yoon Kim for creating the ColabFold logo.
A colab by Sergey Ovchinnikov (@sokrypton), Milot Mirdita (@milot_mirdita) and Martin Steinegger (@thesteinegger).